
Restriction enzymes (REs) have been the workhorses of molecular biology for decades.1 Primarily considered tools of cloning, REs have found a place in some next-generation sequencing techniques that require efficient production of defined DNA ends for subsequent modification and ligation. For example, most flavors of genome-wide chromosome conformation capture use one or two REs to fragment DNA before dilute ligation of fixed chromatin complexes.2,3 Additionally, chromatin accessibility can be inferred by sequencing RE-digested chromatin4,5, though such methods are less popular than those which utilize sequence-agnostic enzymes like DNase-seq6 and ATAC-seq.7 Generally, if you want sticky ends to add adapters or labeled nucleotides, REs are the way to go.
In context of metagenomics, RE-based proximity ligation libraries have been of great utility in improving metagenome-assembled genomes8 and linking plasmids with their bacterial hosts.9,10 When working with a single organism that has a well-annotated genome, sequence bias can be accounted for by choosing an enzyme that is expected to cut at regular intervals across the genome, based on GC content or the known distribution of RE sites. Applying RE methods to a microbiome presents a problem - how do you choose an appropriate enzyme when there are many different genomes, or when the community composition is unknown?

Metagenomic proximity ligation sequencing associates plasmids with bacterial chromosomes, without the need to isolate and culture the host. (Made with BioRender)
To address this question, I looked at the frequencies of RE recognition sites across a collection of human-associated bacterial genomes…
Enzymes
Because this question concerns the choice of REs for metagenomics, I only considered REs that are commercially available and useful in context of the methods described. To that end, I downloaded a list of available enzymes and their recognition sequences from NEB and subset the RE list to exclude (1) enzymes with ambiguous bases at any position in the recognition sequence (the tool I use does not handle complete IUPAC encoding, only ACGT) and (2) enzymes that cleave outside of the recognition site (i.e. Type IIS enzymes). For simplicity, different REs with the same recognition sequence share the same index, even if they are not isoschizomers, yielding 14 unique 4-bp sequences, 60 unique 6-bp sequences (some representing nicking endonucleases), and 9 unique 8-bp sequences. The cleaned-up RE data are available here.
Genomes
The human microbiome is one of the best-studied microbial systems, and the PATRIC database11 maintains a large collection of whole-genome sequencing data for microbes isolated from human hosts. Using a version of this database accessed in 2018, I subset the genomes to exclude those smaller than 1 Mbp and, for simplicity of visualization, included only bacteria from the phyla commonly seen in the human gut microbiome. In total, 59,579 genomes representing 6 phyla were analyzed.
Analysis
I used Jellyfish to count all k-mers of lengths 4, 6, and 8 across the set of genomes. Jellyfish utilizes memory-efficient search of query sequences using hash tables and, importantly, does not double-count palindromes.12 Counts were parsed to extract sequences corresponding to the curated RE set, and I used a simple normalization to account for both genome length and recognition sequence length (n), with an added “0.1” to avoid errors when plotting zero-counts on a log axis:
\[ NormCount = \frac{RawCount + 0.1}{GenomeLength} \times 4^n \] Some caveats:
- This analysis does not account for the presence or activity of methyltransferases in bacterial genomes, and some REs are sensitive to DNA methylation. Consider isoschizomers DpnII, Sau3AI, and MboI: all three enzymes recognize the GATC tetramer and cleave it to produce a CTAG-5’ overhang. However, both DpnII and MboI are blocked by dam methylation, whereas Sau3AI is agnostic to dam methylation state. DpnI, which also recognizes GATC but produces blunt ends, requires dam methylation in order to cleave. While resources like REbase13 provide data on the specificity of methyltransferases across bacteria, predicting the methylation state of bacteria in complex communities is complicated by phase variation,14 though long-read technologies are making meta-epigenomics more feasible.15,16
- Many of the genomes available from PATRIC are incomplete and composed of multiple contigs. This is especially true for the metagenome-assembled genomes, some of which are highly fragmented and composed of hundreds or thousands of contigs. For bacterial chromosomes that exist in PATRIC as fragments, it may be the case that some RE recognition sites occur in the gaps between contigs, though I expect such undercounting to be negligible.
- The number of times a sequence appears in a genome does not tell us the distribution of that sequence. If a sequence appears 5 times in a circular 5 Mb genome, a uniform distribution assumes that the sequence occurs once per Mb. However, a random sequence in a random genome will not be perfectly uniformly distributed, and it has been observed that certain palindromes are more uniform or more clustered than expected by a random model.17
Results
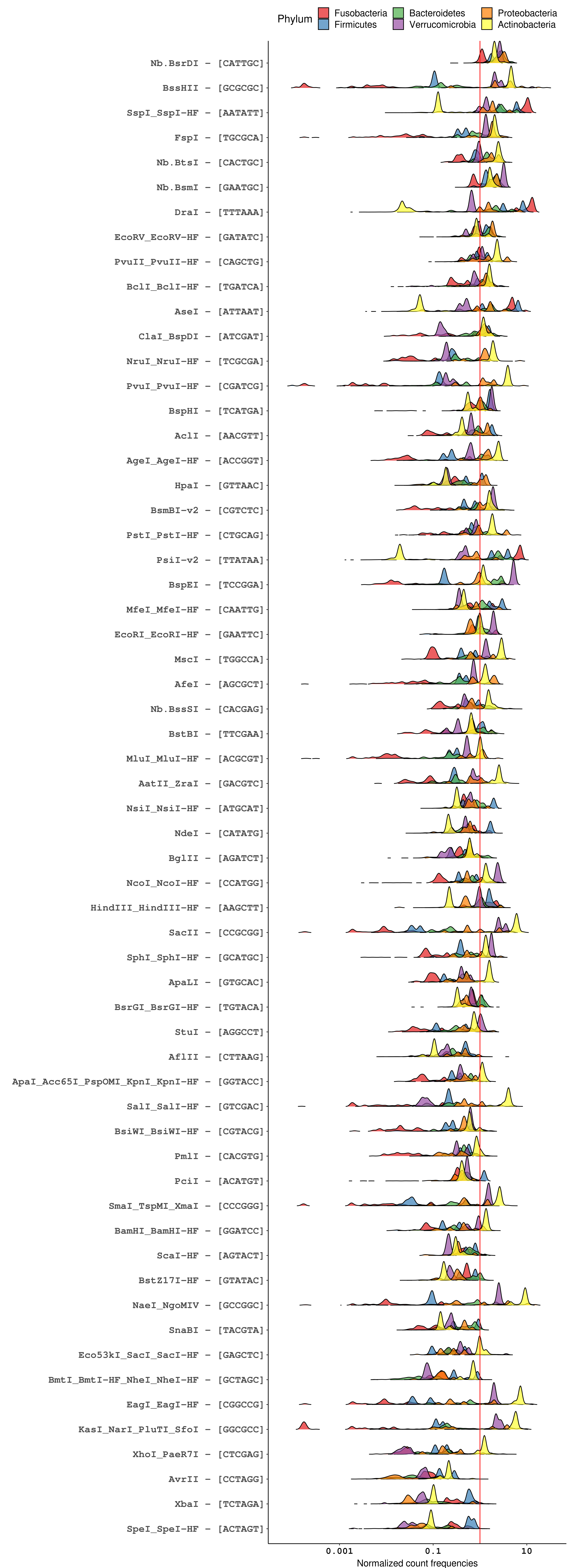
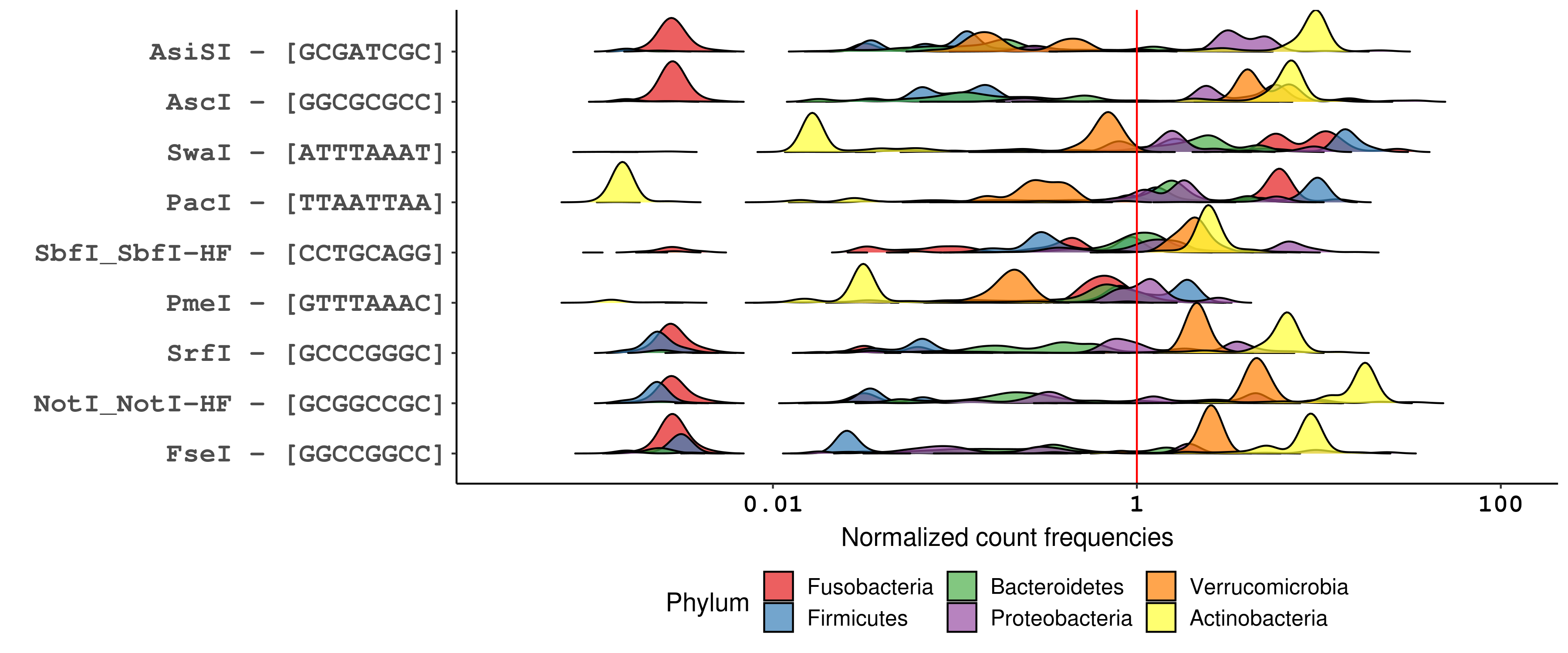
Plotting RE site frequencies distributions for the six chosen phyla, it’s obvious that GC content underlies most of the differences in recognition sequence frequency at the phylum level. For example, looking specifically at 4-bp RE recognition sequences (figure below), GC-skewed recognition sequences like HhaI (GCGC) and BstUI (CGCG) are poorly represented across phyla whose genomes tend to be AT-rich, particularly Firmicutes and Fusobacteria. Likewise, the GC-rich genomes of Actinobacteria are predicted to be infrequently cut by AT-skewed enzymes like MseI (TTAA) and MluCI (AATT). Similar trends can be seen for enzymes with 6-bp and 8-bp recognition sequences.
{kind=link}
{kind=link}

Distribution of normalized recognition sequence frequencies for commercially available NEB 4-cutters across bacterial phyla of the human microbiome. The red line marks a normalized count frequency of 1, which represents the frequency at which a random 4-mer is expected to occur in a random genome, on average (1 occurence per 4^4 bp). Enzymes are ordered top-to-bottom by the median normalized count frequency across all genomes.
However, choosing a RE based solely on GC concordance between genomes and recognition sequences does not account for the biology of specific sequences. An interesting case is that of BfaI, which recognizes the CTAG tetramer. Despite having the same recognition sequence base composition as frequent-cutting enzymes like HpyCH4V (TGCA) and CviAII (CATG), BfaI is the least frequent 4-cutter across the bacterial genomes analyzed. Similar infrequency is seen for the 6-bp cutters that share a core CTAG tetramer, namely AvrII (CCTAGG), XbaI (TCTAGA), SpeI (ACTAGT), and NheI (GCTAGC). The relative rarity of CTAG is a trait conserved across bacterial genomes, and its specific depletion in protein-coding genes makes CTAG useful as a marker of intergenic regions.18 Biologically, the depletion of CTAG in bacterial genomes is hypothesized to be related to the use of rare codons as a mechanism of global gene expression regulation19,20 and may be consequence of sequence bias in VSP repair.21,22
So, what can we take from all this? For metagenomic proximity ligation experiments, particularly experiments involving human microbiome samples, there are several REs that are minimally biased, though no option is perfect. Concerning frequent-cutting enzymes with 4-bp recognition sequences, both HpyCH4V and AluI are evenly represented across diverse bacterial phyla, with the added benefit that the activities of both enzymes are unaffected by dam and dcm methylation. However, both HpyCH4V and AluI generate blunt ends, precluding these REs from use in experiments where sticky ends are needed for biotinylated dNTP incorporation.
| HpyCH4V | AluI |
|---|---|

|

|
Among REs with 6-bp recognition sequences, EcoRI and BspHI appear to be the best choices for enzymes with minimal phyla-level bias in cut frequency. Additionally, both of these enzymes generate 5’ overhangs that are amenable to polymerase incorporation of labeled nucleotides, though BspHI cutting is impaired by overlapping dam methylation.
| EcoRI | BspHI |
|---|---|

|

|
References
- Loenen WA, Dryden DT, Raleigh EA, Wilson GG, Murray NE. Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res. 2014 Jan;42(1):3-19.
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009 Oct 9;326(5950):289-93.
- Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, Haussler D, Rokhsar DS, Green RE. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016 Mar;26(3):342-50.
- Chen PB, Zhu LJ, Hainer SJ, McCannell KN, Fazzio TG. Unbiased chromatin accessibility profiling by RED-seq uncovers unique features of nucleosome variants in vivo. BMC Genomics. 2014 Dec 15;15(1):1104.
- Chandradoss KR, Guthikonda PK, Kethavath S, Dass M, Singh H, Nayak R, Kurukuti S, Sandhu KS. Biased visibility in Hi-C datasets marks dynamically regulated condensed and decondensed chromatin states genome-wide. BMC Genomics. 2020 Feb 22;21(1):175.
- Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010 Feb;2010(2):pdb.prot5384.
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015 Jan 5;109:21.29.1-21.29.9.
- DeMaere MZ, Darling AE. bin3C: exploiting Hi-C sequencing data to accurately resolve metagenome-assembled genomes. Genome Biol. 2019 Feb 26;20(1):46.
- Stalder T, Press MO, Sullivan S, Liachko I, Top EM. Linking the resistome and plasmidome to the microbiome. ISME J. 2019 Oct;13(10):2437-2446.
- Kent AG, Vill AC, Shi Q, Satlin MJ, Brito IL. Widespread transfer of mobile antibiotic resistance genes within individual gut microbiomes revealed through bacterial Hi-C. Nat Commun. 2020 Sep 1;11(1):4379.
- Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, Machi D, Mao C, Nordberg EK, Olson R, Overbeek R, Pusch GD, Shukla M, Schulman J, Stevens RL, Sullivan DE, Vonstein V, Warren A, Will R, Wilson MJ, Yoo HS, Zhang C, Zhang Y, Sobral BW. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014 Jan;42(Database issue):D581-91.
- Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011 Mar 15;27(6):764-70.
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015 Jan;43(Database issue):D298-9.
- Morovic W, Budinoff CR. Epigenetics: A New Frontier in Probiotic Research. Trends Microbiol. 2020 May 11:S0966-842X(20)30101-3.
- Beaulaurier J, Zhu S, Deikus G, Mogno I, Zhang XS, Davis-Richardson A, Canepa R, Triplett EW, Faith JJ, Sebra R, Schadt EE, Fang G. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat Biotechnol. 2018 Jan;36(1):61-69.
- Beaulaurier J, Schadt EE, Fang G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat Rev Genet. 2019 Mar;20(3):157-172.
- Karlin S, Burge C, Campbell AM. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 1992 Mar 25;20(6):1363-70.
- Tang L, Zhu S, Mastriani E, Fang X, Zhou YJ, Li YG, Johnston RN, Guo Z, Liu GR, Liu SL. Conserved intergenic sequences revealed by CTAG-profiling in Salmonella: thermodynamic modeling for function prediction. Sci Rep. 2017 Mar 6;7:43565.
- Chen GT, Inouye M. Role of the AGA/AGG codons, the rarest codons in global gene expression in Escherichia coli. Genes Dev. 1994 Nov 1;8(21):2641-52.
- Blattner FR, Burland V, Plunkett G 3rd, Sofia HJ, Daniels DL. Analysis of the Escherichia coli genome. IV. DNA sequence of the region from 89.2 to 92.8 minutes. Nucleic Acids Res. 1993 Nov 25;21(23):5408-17.
- Merkl R, Kröger M, Rice P, Fritz HJ. Statistical evaluation and biological interpretation of non-random abundance in the E. coli K-12 genome of tetra- and pentanucleotide sequences related to VSP DNA mismatch repair. Nucleic Acids Res. 1992 Apr 11;20(7):1657-62.
- Gutiérrez G, Casadesús J, Oliver JL, Marín A. Compositional heterogeneity of the Escherichia coli genome: a role for VSP repair? J Mol Evol. 1994 Oct;39(4):340-6.